Imagine that you are a waiter running back and forth in your breakfast restaurant. Your life is constantly moving between the kitchen and the seating area in your usual “flow”. Most days you have to work very hard to make ends meet, so you don’t have time to sit back and smell the roses or rose the smells. It’s a shame that your work prevents you from studying the world around you through mathematics and algorithms. Every now and then, a guest orders a stack of pancakes, but, when the cook hands you the plate of pancakes, you’re a bit disappointed because the pancakes aren’t stacked by size.
 |
| what is this madness |
What self-respecting philopancakist would tolerate such blasphemy? The proper pancake stack must place the largest pancake on bottom, the smallest pancake on top, and fill the space with the pancakes in ascending order. This is the only way you can pour syrup on it so that the syrup touches each pancake. It should be the responsibility of the cook to flip the pancakes in such a way that lines them up from largest on bottom to smallest to top with everything making sense in between.
This begs the question, if someone gives you a randomly assorted stack of pancakes, how do you sort them through flipping them? Namely, what’s the most efficient way for us to look at a set of random numbers and sort them from least to greatest (or vice versa) by reversing different segments of those numbers? This is the Pancake-Sort problem, and the number of flips is known as the reversal distance.
A man named Bill Gates proposed a solution to the Pancake-Sort problem. You can read it here.
What makes this problem more interesting is that it has application in biology in the study of genetic inversions. DNA bases experience a type of mutation known as inversions in which segments of bases are reversed. This can occur with a small segments of genes or multiple genes.
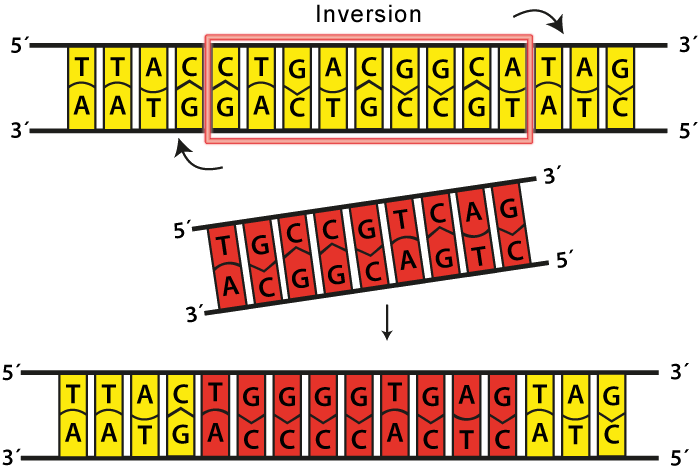
We want to know how many inversions that a certain gene or region of the genome has undergone because that tells us how old the DNA is or how much it has evolved. Two species that share a large reversal distance may have evolved farther apart than two that share similar reversal distances.
Perhaps it is ironic to mention that mother nature’s love for making molecular biological interactions actually makes this problem much easier to solve. In biology, we don’t have more than four different DNA bases, and our bases are actually aligned in such a way that there is a “forward” and “reverse” direction to each string. This means that each base must be aligned in the forward or reverse direction in order for that string to function properly. Taking these into account will make the problem simpler because we can restrict ourselves to aligning the DNA strings so that these conditions are satisfied.
When I first approached a simple version of this problem, I wrote a solution that would take the input string that needs to be sorted and judge potential inversions by their hamming distance from the desired end sequence. By constantly following the potential inversion that had the lowest Hamming distance, we would hope to find the end result. (Hamming Distance is the number of bases between two strings that do not match when the two are aligned. So “AAAG” and “AAAA” would have a hamming distance of 1 since they differ by one base.) Basically, this approach would try to find the shortest way to get from the beginning to the end by seeing which inversion would match the end result the most, and repeating this process until the end result is reached. But, even intuitively, this approach would not necessarily find the end result in all scenarios. It may end up creating loops and traversing through inversions that would have low hamming distances but not move in the most optimal path from the first string to the end. (You can see some of my solution here.)
