People who are just beginning to code make a lot of mistakes and do a lot of stupid things. I once used to struggle with parsing thorough every line of a file before I learned that it would be much easier to use split-functions and similar lists. One common mistake people make is that they need to store large amounts of data and parse through them every time they need to access that information. But, with a bit more expertise, those newbies can throw away those “for” loops and sort-methods. There’s a new kid in town. And his name is “Hashing.”
for i in range(len(s)):
c+=1
if c in indices:
line=int(indices_start.index(c))
for i in s[i:indices_end[line]+1]:
s2+=“N”
The problem is that this takes forever and a half to run. But, if we just replaced the two lists with a single hash (or dictionary) which we will name indices, then the program runs like clockwork.
for i in range(len(s)):
c+=1
if c in indices:
line=int(indices[c])
for i in s[i:line+1]:
s2+=“N”
Just goes to show how much of a difference code optimization can make.
END
(Excuse me for that last line. My Fortran is leaking.)
Perl may be crafty and efficient like a ninja, Ruby may be written like a prose or work of fiction, but, for most purposes, Python, with its simplicity and elegance, is usually my weapon of choice when it comes to programming languages. (To be frank, as long as it’s not some cryptic code like Fortran that should probably be waiting for the rain to wash it away, it floats my boat.) With my knack for mathematics, I had been reconstructing various equations and theorems from scratch in most of my scripts. Recently, I’ve begun to embrace NumPy to give me more functionality for purposes like matrices and arrays, but also that I can do all the things my MATLAB friends do without too much effort to learn extra languages.
(In fact, while we’re at it, let’s just put everything in Python! Python-Excel, Python-sql, import everything!)
Back in my pancake post, I talked about how you can use a simple two-step algorithm for sorting out a string of numbers. In this post, however, I’m going to talk about sorting through two different string of letters to find their longest common subsequence. Unlike the challenge of the longest common substring, the subsequence need not consist solely of letters that are adjacent to one another, but can contain letters separated. So, this means that the longest common subsequence between “AACTTG” and “ACTGG” would be not be “ACT”, but “ACTG”.
I found this problem interesting because it gave me the chance to flex my NumPy muscles and look for more “indirect” ways of solving a problem rather than using a brute-force Ctrl+F-esque approach that wipes away your entire RAM when you search through strings longer than 30 characters.
Fret not! For we can construct a matrix of some sort to help us with this issue. In bioinformatics, we can solve this problem using a scoring matrix. By taking advantage of the set of possible DNA bases {A, C, T, G}:
Simply place your two DNA strings on the axes and move each number in the grid from the top-left to bottom-right . If there is a match in the base between the two strings at a certain location, add one to the number. Then follow your path form highest to lowest value. (source)
This is known as the Traceback approach, and we can optimize it further with hashes for lengths and in other ways.
I wrote a solution from this method (drawing heavily upon other sources) to solve this problem here, although I’m still fixing up some issues in it from converting between different syntaxes and formats.
One of my current projects in the Matthew Hahn Lab is to investigate the effectiveness of a few different full-genome alignment methods. My mentor and I have been studying a new program called progressiveCactus, and comparing its output to other alignment methods. By comparing the number of indels (that is, insertions and deletions) between different species, we can compare the effectiveness of different genome-alignment methods. But my work has mostly been spent struggling to figure out how to get programs to run, and deciding the best way to parse output files.
How does progressiveCactus work, you ask? When I tried to answer that question the moment I began working in the Hahn Lab, I couldn’t figure a thing out. After gaining much more experience in bioinformatics and analysis of complex systems, though, it has made more sense to me.
In order to allow multiple genomes to align to one another in any possible way, we can arrange them in a circular pattern, as shown above. This lets us create threads of different colors, in which each color represents a different sequence. The ends of the boxes (A1 and A4) are the telomeres, as in, the ends of the chromosomes. It’s easy to find reverse complements, similarities, and other neat features. When we combine all of the different circular genome plots, this way, we can create “cactus” graphs.
Pictured: a cactus
From these chains, we can create entire networks upon networks to give us full-aligned genomes. progressiveMauve has been shown to be very quick and effective with a small number of different genomes, and it has a very attractive GUI, as well. We’re focusing on the output from this program to compare to that of progressiveCactus.
Ever since I finished my work at Cornell, I’ve been much more confident and focused in my research at IU. I look forward to continuously keep moving onto bigger and better things in research and elsewhere.
Imagine that you are a waiter running back and forth in your breakfast restaurant. Your life is constantly moving between the kitchen and the seating area in your usual “flow”. Most days you have to work very hard to make ends meet, so you don’t have time to sit back and smell the roses or rose the smells. It’s a shame that your work prevents you from studying the world around you through mathematics and algorithms. Every now and then, a guest orders a stack of pancakes, but, when the cook hands you the plate of pancakes, you’re a bit disappointed because the pancakes aren’t stacked by size.
what is this madness
What self-respecting philopancakist would tolerate such blasphemy? The proper pancake stack must place the largest pancake on bottom, the smallest pancake on top, and fill the space with the pancakes in ascending order. This is the only way you can pour syrup on it so that the syrup touches each pancake. It should be the responsibility of the cook to flip the pancakes in such a way that lines them up from largest on bottom to smallest to top with everything making sense in between.
This begs the question, if someone gives you a randomly assorted stack of pancakes, how do you sort them through flipping them? Namely, what’s the most efficient way for us to look at a set of random numbers and sort them from least to greatest (or vice versa) by reversing different segments of those numbers? This is the Pancake-Sort problem, and the number of flips is known as the reversal distance.
A man named Bill Gates proposed a solution to the Pancake-Sort problem. You can read it here.
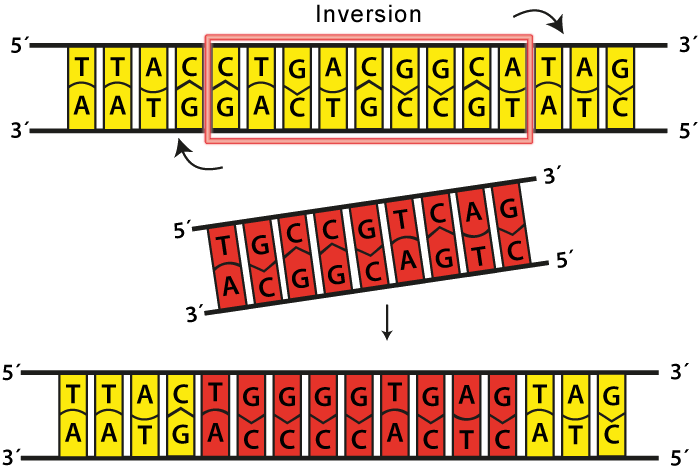
What makes this problem more interesting is that it has application in biology in the study of genetic inversions. DNA bases experience a type of mutation known as inversions in which segments of bases are reversed. This can occur with a small segments of genes or multiple genes.
We want to know how many inversions that a certain gene or region of the genome has undergone because that tells us how old the DNA is or how much it has evolved. Two species that share a large reversal distance may have evolved farther apart than two that share similar reversal distances.
Perhaps it is ironic to mention that mother nature’s love for making molecular biological interactions actually makes this problem much easier to solve. In biology, we don’t have more than four different DNA bases, and our bases are actually aligned in such a way that there is a “forward” and “reverse” direction to each string. This means that each base must be aligned in the forward or reverse direction in order for that string to function properly. Taking these into account will make the problem simpler because we can restrict ourselves to aligning the DNA strings so that these conditions are satisfied.
When I first approached a simple version of this problem, I wrote a solution that would take the input string that needs to be sorted and judge potential inversions by their hamming distance from the desired end sequence. By constantly following the potential inversion that had the lowest Hamming distance, we would hope to find the end result. (Hamming Distance is the number of bases between two strings that do not match when the two are aligned. So “AAAG” and “AAAA” would have a hamming distance of 1 since they differ by one base.) Basically, this approach would try to find the shortest way to get from the beginning to the end by seeing which inversion would match the end result the most, and repeating this process until the end result is reached. But, even intuitively, this approach would not necessarily find the end result in all scenarios. It may end up creating loops and traversing through inversions that would have low hamming distances but not move in the most optimal path from the first string to the end. (You can see some of my solution here.)
This new office at Indiana University is actually keeping in touch with me to promote research opportunities for other students at my university.
To give a brief background, when I entered Indiana University, I was so obsessed with science that I was almost desperate to join a research lab. After emailing around a few professors, I was offered spot in the Matthew Hahn Lab to study Bioinformatics. Soon enough, I helped a few of my friends get into labs, too, by giving them advice and instructions about how I did it. Later, during my freshman year, I was accepted to a full-time summer internship at Cornell University that paid for transportation, housing, food, and a gave a $5000 stipend. From all of these experiences, I’ve compiled my advice and instructions into a guide from the beginning to the end.
If you’re a college student reading this blog and looking for research opportunities at your university or advice and tips about undergraduate research or you just want to read more about my experience, check out my “Scientific Research Guide” under “My Work” over there on the right.
As for now, I’m currently working on a new approach to my project in my lab.